Applying single-cell sequencing approaches to characterize the healthy and malignant bone marrow microenvironment
Leukemia is a clonal hematopoietic neoplasm characterized by the proliferation and accumulation of lymphoid or myeloid progenitor cells throughout the bone marrow. Extensive genomic characterization uncovered multiple candidates for targeted therapy. Unfortunately, most targeted therapies fail to elicit prolonged disease remission due to the emergence of pre-existent or de novo therapy-resistant leukemic clones. There is compelling emerging evidence that cell non-autonomous contributions to leukemia play a pivotal role in disease development, propagation, and maintenance. One has to consider the various cellular components of the bone marrow microenvironment that form among them a network of molecular interactions. This array of cell types includes immune cells, adipocytes, bone-forming osteoblasts, mesenchymal stromal cells, and vascular endothelial cells. Numerous studies support a role of the microenvironment in maintenance of the leukemic cells as well as treatment resistance.
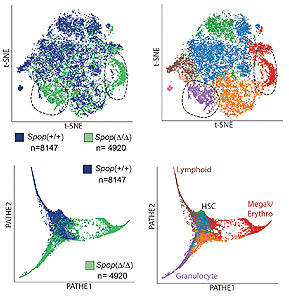
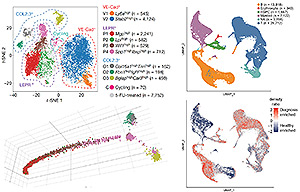
To better understand the function, heterogeneity, and the complexity of the bone marrow microenvironment in normal hematopoiesis and leukemia, we are using a combination of single-cell assays, including scRNA-Seq, CITE-Seq, and scATAC-Seq, in combination with computational methods to generate insights into this high-resolution biological data. Our work (Tikhonova, Dolgalev et al. Nature 2019) has characterized a previously unappreciated level of cellular heterogeneity within the bone marrow stromal niche, identified novel cellular subsets, reconstructed developmental trajectories, and resolved cellular sources of pro-hematopoietic growth factors, chemokines, and membrane-bound ligands. Under conditions of stress, our studies revealed a transcriptional remodeling of these niche elements accompanying the myeloid skewing that characterizes emergency hematopoiesis. To investigate the role of the microenvironment in B cell acute lymphoblastic leukemia (B-ALL) progression and treatment evasion, we utilized single-cell approaches to generate a comprehensive map of the primary human bone marrow immune microenvironment throughout three distinct stages of the disease: diagnosis, remission and relapse (Witkowski, Dolgalev et al. Cancer Cell 2020). We showed extensive re-modeling of the immune microenvironment composition throughout the course of conventional chemotherapy and uncover a role for leukemia-associated non-classical monocytes in promoting B-ALL pathogenesis in vivo. We took advantage of CITE-seq to expand the single-cell measurements beyond the transcriptome and simultaneously profile surface proteins, further validating our findings. Out studies provide a greater understanding of the potential extrinsic regulators of B-ALL survival and may highlight previously unknown environmental factors influencing immune-based treatment approaches to high-risk B-ALL.